

Charakterystyka dystrofiny, struktura i funkcje
- 2886
- 313
- Eugenia Czapla
dystrofina Jest to białko w kształcie trzciny lub pręta związanego z błoną komórek mięśni szkieletowych, gładkich i sercowych, również prezentuje komórki nerwowe i w innych narządach ludzkiego ciała.
Ma funkcje podobne do innych białek cytoszkieletu i uważa się, że działa głównie w stabilności błony włókien mięśniowych i w połączeniu zewnątrzkomórkowej błony podstawnej z wewnątrzkomórkowym cytoszkieletem.
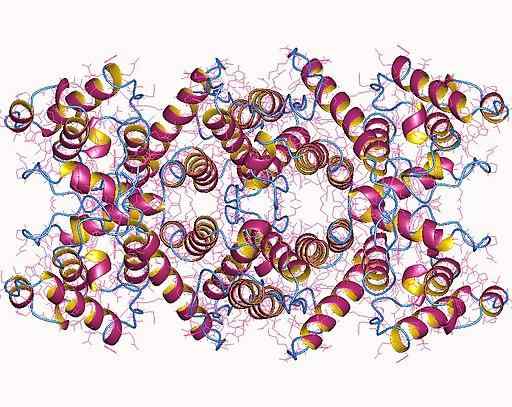
Struktura molekularna dystrofiny (źródło: Norwood, F.L., Sutherland-Smith, a.J., Keep, n.H., Kendrick-Jones, J.; Autor wizualizacji: Użytkownik: AstroJan [CC BY-SA 4.0 (https: // creativeCommons.Org/licencje/nabrzeże/4.0)] przez Wikimedia Commons) Jest kodowany na chromosomie X, w jednym z największych genów opisanych dla ludzi, z których niektóre mutacje są zaangażowane w patologie związane z chromosomami płciowymi, jako dystrofia mięśni Duchenne'a (DMD) (DMD) (DMD).
Ta patologia jest drugim najczęstszym odziedziczonym zaburzeniem na świecie. Wpływa na jednego na 3500 mężczyzn, co staje się widoczne od 3 do 5 lat jako przyspieszone zużycie mięśni, które może skrócić czas życia do nie więcej niż 20 lat.
Gen dystrofiny został po raz pierwszy wyizolowany w 1986 roku i scharakteryzowano za pomocą klonowania pozycyjnego, co oznaczało duży postęp dla genetyki molekularnej tamtych czasów.
[TOC]
Charakterystyka
Dystrofina jest bardzo zróżnicowanym białkiem związanym z błoną plazmatyczną komórek mięśniowych (sarkoma) i innych komórek różnych układów ciała.
Jego różnorodność wynika z procesów związanych z regulacją ekspresji genu, który go koduje, który jest jednym z największych genów opisanych dla ludzi. To dlatego, że ma więcej niż 2.5 milionów par zasad, reprezentujących około 0.1% genomu.
Ten gen. Składa się z około 99% intronów, a region kodujący jest reprezentowany tylko w 86 eksonach.
Może ci służyć: czerwony fenol: cechy, przygotowanie, zastosowaniaRozpoznawane są trzy różne izoformy tego białka, które pochodzą z tłumaczenia posłańców, które są transkrybowane z trzech różnych promotorów: takie, które występują tylko w neuronach korowych i hipokampowych, kolejnych w komórkach Purkinjego (również w mózgu), a ostatni w mięśniach mięśniowych Komórki (szkieletowe i sercowe).
Struktura
Ponieważ gen dystrofiny można „odczytać” z różnych wewnętrznych promotorów, istnieją różne izoformy tego białka, które oczywiście mają różne rozmiary. Na tej podstawie struktura „kompletnych” i „krótkich” izoform jest opisana poniżej.
„Whole” lub „kompletne” izoformy
„Całe” izoformy dystrofiny to białka w kształcie trzciny, które mają cztery podstawowe domeny (N-końcowa, domena centralna, domena bogata w cystein i domena C-końcowa), które razem ważą nieco ponad 420 kDa i mają więcej lub mniej 3 3.685 odpady aminokwasowe.
Domena N-końcowa jest podobna do α-aktyniny (białko związane z aktyną) i może mieć między 232 a 240 aminokwasami, w zależności od izoformu. Domena centralna lub trzcinowa składa się z 25 potrójnych powtarzanych powtarzanych, podobnych do spektrii i ma około 3000 odpadów aminoacidalnych.
C-końcowy region domeny centralnej, która jest tworzona przez bogatego bogatego w cysteinę, ma około 280 odpadów i jest bardzo podobny do przyczyny unia wapnia obecnego w białkach takich jak kalmodulina, α-aktynana i β β-spectrine. C-końcowa domena białka składa się z 420 aminokwasów.
„Krótkie” izoforms
Ponieważ gen dystrofiny ma co najmniej cztery wewnętrzne promotory, mogą występować białka o różnych długościach, które różnią się od siebie przez brak którejkolwiek z ich domen.
Każdy z wewnętrznych promotorów ma wyjątkowy poprzedni egzonegent), który jest wyrażany w różnych regionach ciała.
Może ci służyć: hormony steroidowe: struktura, synteza, mechanizm działaniaDP260 jest wyrażany w siatkówce i współistnieje z „kompletnymi” formami mięśni i mózgu. DP140 znajduje się w mózgu, w siatkówce i w nerkach, podczas gdy DP116 występuje tylko w nerkach obwodowych dorosłych, a DP71 jest w większości tkanek nie -muzcle.
Funkcje
Według różnych autorów dystrofina ma różne funkcje, które nie tylko sugerują jej udział jako białko cytoszkieletu.
Stabilność błony
Główną funkcją dystrofiny, jako cząsteczki związanej z błoną nerwową i mięśniową, jest oddziaływanie z co najmniej sześcioma różnymi kompleksowymi białkami błonowymi, z którymi łączy się, tworząc kompleksy dystropiny-glukoproteiny.
Tworzenie tego kompleksu generuje „most” przez błonę komórkową mięśni lub sarkolemy i łączy „elastycznie” bazę macierzy zewnątrzkomórkowej z wewnętrznym cytoszkieletem.
Kompleks dystrofiny-glukoprotein działa w stabilizacji błony i ochronie włókien mięśniowych przed martwicą lub uszkodzeniem spowodowanym przez indukowane skurczu przez długi czas, co zostało wykazane przez odwrotną genetykę.
Ta „stabilizacja” jest zwykle postrzegana jako analog.
Transdukcja sygnału
Dystrofina lub raczej kompleks białkowy, który tworzy się z glikoproteinami w błonie, nie tylko wykonuje funkcje strukturalne, ale również wskazywało, że może mieć pewne funkcje w sygnalizacji komórkowej i komunikacji.
Jego lokalizacja sugeruje, że może uczestniczyć w przenoszeniu napięcia z włókien aktyny w sarkomerach włókien mięśniowych przez błonę plazmatyczną w kierunku matrycy pozakomórkowej, ponieważ jest fizycznie związana z tymi włóknami i przestrzenią zewnątrzkomórkową przestrzeni zewnątrzkomórkowej.
Może ci służyć: jasnozielony agar: co to jest, podkład, przygotowanie, użyciaDowody innych funkcji w transdukcji sygnału zostały oderwane od niektórych badań przeprowadzonych z mutantami dla genu dystrofiny, w których wady obserwuje się w wodospadach sygnalizacyjnych, które mają związek z zaprogramowaną śmiercią komórki lub obroną komórki.
Bibliografia
- Ahn, a., & Kunkel, L. (1993). Różnorodność strukturalna i funkcjonalna dystrofiny. Nature Genetics, 3, 283-291.
- Doubek, r. W. (1950). Histologia o wysokiej wydajności (2 wyd.). Philadelphia, Pensylwania: Lippinott Williams & Wilkins.
- Ervasti, J., I Campbell, K. (1993). Dystrofina i szkielet membranowy. Obecna opinia w biologii komórkowej, 5, 85-87.
- Hoffman, e. P., Brown, r. H., & Kunkel, L. M. (1987). Dystrofina: produkt białkowy locus dystrofii mięśniowej Duchenne. Komórka, 51, 919-928.
- Koenig, m., Monako, a., & Kunkel, L. (1988). Pełny cytoszkielet w kształcie pręta sekwencji białka dystrofiny przewiduje A. Komórka, 53, 219-228.
- Czytać., Winder, s. J., & Hubert, j. (2010). Biochimica et biophysica akta dystrofina: więcej niż suma jej części. Biochimica et biophysica acta, 1804(9), 1713-1722.
- Miłość, d., BYTH, ur., Tensley, J., Blake, d., & Davies, k. (1993). Dystrofina i białka związane z dystrofiną: przegląd badań białka i RNA. Neuromuch. Disord., 3(1), 5-21.
- Muntoni, f., Torelli, s., & Ferlini, do. (2003). Dystrofina i mutacje: jeden gen, kilka białek, wiele fenotypów. Neurologia Lancet, 2, 731-740.
- Pasternak, c., Wong, s., I Elson, E. L. (1995). Mechaniczna funkcja dystrofiny w komórkach mięśniowych. Journal of Cell Biology, 128(3), 355-361.
- Sadoulet-Puccio, godz. M., & Kunkell, L. M. (1996). Dystrofina i jej lsoformy. Patologia mózgu, 6, 25-35.
- « Charakterystyka oksymoglobiny, struktura i krzywa połączenia
- Charakterystyka, struktura i funkcje tropomiozyny »