

Struktura glikogenu, synteza, degradacja, funkcje
- 2031
- 93
- Maksymilian Kępa
On Glikogen Jest to węglowodan do przechowywania większości ssaków. Węglowodany są powszechnie nazywane cukrami i są one klasyfikowane zgodnie z liczbą odpadów spowodowanych przez hydrolizę (monosacharydy, disacharydy, oligosacharydy i polisacharydy)))).
Monosacharydy to najprostsze węglowodany, które są klasyfikowane zgodnie z liczbą węgli zawartych w ich strukturze. Wówczas są trio (3c), tetrosas (4c), pentosas (5c), sześciokosowy (6c), heptosaza (7c) i oktosas (8c).
Chemiczna struktura glikogenu wykazująca wiązania glikozydowe (źródło: glykogen.SVG: Neuropoger Pochodna Praca: Marek M [domena publiczna] za pośrednictwem Wikimedia Commons) Zgodnie z obecnością grupy aldehydu lub grupy Cetona, te monosacharydy są również klasyfikowane odpowiednio jako Aldies lub Ketosas.
Disacharydy powodują hydrolizę, dwa proste monosacharydy, podczas gdy oligosacharydy wytwarzają 2 do 10 jednostek monosacharydów i polisacharydów wytwarzają więcej niż 10 monosacharydów.
Glikogen wynika z biochemicznego punktu widzenia, polisacharyd złożony z rozgałęzionych łańcuchów sześciokrębowej aldozy, czyli heksozy znanej jako glukoza. Graficznie może być reprezentowany na glikogen jako drzewo glukozy. Nazywa się to również skrobią zwierząt.
Glukoza w roślinach jest przechowywana jako skrobia i u zwierząt jako glikogen, który jest przechowywany głównie w wątrobie i tkance mięśniowej.
W wątrobie glikogen może ustalić 10% swojej masy i 1% masy mięśniowej. Podobnie jak u mężczyzny o długości 70 kg, wątroba waży około 1800 g, a mięśnie około 35 kg, całkowita ilość glikogenu mięśni jest znacznie większa niż wątroba.
[TOC]
Struktura
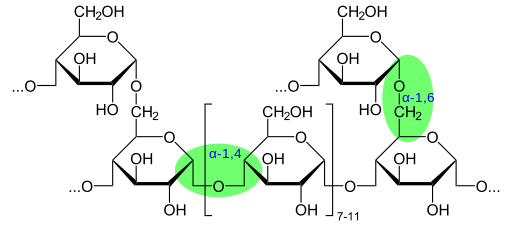
Masa cząsteczkowa glikogenu może osiągnąć 108 g/mol, równoważne cząsteczkom glukozy 6 x 105. Glikogen składa się z wielu rozgałęzionych łańcuchów α-D-glikozy. Glukoza (C6H12O6) jest aldohexosa, który może być reprezentowany w sposób liniowy lub cykliczny.
Glikogen ma bardzo rozgałęzioną i zwartą strukturę z łańcuchami 12 do 14 odpadów glukozy w postaci α-D-glukozy, które są powiązane z wiązaniami glukozydowymi α- (1 → 4). Konsekwencje łańcucha są tworzone przez α- (1 → 6) glukozydowe.
Glikogen, podobnie jak skrobia spożywana w diecie, zapewnia większość węglowodanów, których potrzebuje ciało. W jelicie te polisacharydy są degradowane przez hydrolizę, a następnie wchłaniane w kierunku potoku krążenia głównie jako glukoza.
Trzy enzymy: ß-amylaza, α-amylaza i amylo-α- (1 → 6)-glukozydaza są odpowiedzialne za degradację jelit zarówno glikogenu, jak i skrobi.
Α-amylaza losowo hydrolizuje wiązania α (1 → 4) łańcuchów bocznych zarówno glikogenu, jak i skrobi, a zatem otrzymuje nazwę endoglikysidazy. Ss-amyla jest egzoglikozydazą, która uwalnia ß-maltozę dímeros łamie α- (1 → 4) glikozydowe z końców najbardziej zewnętrznych łańcuchów bez dotarcia do konsekwencji.
W związku z faktem, że ani ß-amylaza, ani α-amylazy nie degradują gałęzi, końcowy produkt jej działania nie jest wysoce rozgałęziona struktura około 35 do 40 reszt, które są nazywane granicą dextrine.
Limit dekstryny jest ostatecznie hydrolizowany w punktach rozgałęzienia, które mają wiązania α- (1 → 6) przez amyle-α- (1 → 6)-glukozydazę, znaną również jako „zniesławiający” enzym. Łańcuchy uwalniane przez ten defloaż są po zdegradowaniu przez ß-amylazę i α-amylazę.
Ponieważ połknięty glikogen wchodzi jako glukoza, ten występujący w tkankach musi być syntetyzowany przez organizm z glukozy.
Może ci służyć: puryny: charakterystyka, struktura, funkcjeSynteza
Synteza glikogenu nazywa się glikogenezą i odbywa się szczególnie w mięśniu i wątrobie. Glukoza wchodząca do organizmu z dietą przechodzi do torrentu krążenia i stamtąd wewnątrz komórek, gdzie jest natychmiast fosforylowana przez enzym zwany glikoquinazą.
Glukochinaza fosforylowa do glukozy w węglu 6. ATP zapewnia fosfor i energię do tej reakcji. W rezultacie powstaje glukoza 6-fosforan i uwalnia się ADP. Następnie glukoza 6-fosforanowa staje się 1-fosforanem glukozy przez działanie fosfoglukumutazy, która błotnia fosfor od pozycji 6 do pozycji 1.
1-fosforanowa glukoza jest aktywowana do syntezy glikogenu, która implikuje uczestnictwo zestawu trzech innych enzymów: pirofosforyazę UDP-glikozę, syntetycznym glikogenu i amilo- (1,4 → 1,6)-gicosyltransferazy.
Glukozo-1-fosforan wraz z trifosforanem urydyny (UTP, nukleozyd tridyny tridyny) i działaniem UDP-glikosfosforylaza, tworzą kompleks difosforan-glukozę urydyny (UDP GLC) (UDP GLC). W trakcie procesu jon pirofosforanowy jest hydrolizowany.
Następnie syntetyczny enzym glikogenu tworzy wiązanie glukozydowe między C1 kompleksu GLC UDP a C4 końcowej pozostałości glikogatu glikogenu, a UDP UDP jest uwalniany z kompleksu glukozowego aktywowanego UDP. Aby ta reakcja wystąpiła, musi istnieć wstępna cząsteczka glikogenu zwana „pierwotnym glikogenem”.
Pierwotny glikogen jest syntetyzowany na białku gruntownym, glikogeninie, które ma 37 kDa i Glysila w pozostałości tyrozynowej za pomocą kompleksu UDP GLC. Stamtąd są powiązane odpady α-D-glukozy z linkami 1 → 4 i powstaje mały łańcuch, na którym działa glikogen syntezazy.
Gdy początkowy łańcuch łączy co najmniej 11 reszt glukozy, enzym rozgałęziony lub amile (1,4 → 1,6) -Glicoslotransferaza przenosi kawałek łańcucha 6 lub 7 odpadów glukozy do sąsiedniego łańcucha w pozycji 1 → 6, który ustanawia odgałęzienie punkt. W ten sposób zbudowana cząsteczka glikogenu rośnie przez dodanie jednostek glukozy o łączach glikozydowych 1 → 4 i więcej konsekwencji.
Degradacja
Degradacja glikogenu nazywa się glukogenezą i nie jest równoważna odwrotnej ścieżce jej syntezy. Prędkość tej trasy jest ograniczona prędkością reakcji katalizowanej przez glikogen fosforylazy.
Glikogen fosforylazy jest odpowiedzialny za podział (fosforoliza) łączy 1 → 4 z łańcuchów glikogenu, uwalniając 1-fosforan glukozy. Działanie enzymatyczne rozpoczyna się na końcach najbardziej zewnętrznych łańcuchów i są usunięte sekwencyjnie, aż 4 pozostałości glukozy pozostaną po każdej stronie konsekwencji.
Następnie inny enzym, α- (1 → 4) → α- (1 → 4) transfer Glucano, pozostawia punkt odgałęzienia odsłonięty przez przeniesienie jednostki trisacharydowej z jednej gałęzi do drugiej. To pozwala na hydrolys amilo- (1 → 6) -glukozydaza (enzym nienarmowy). Połączone działanie tych enzymów kończy się całkowicie dzielącym się na glikogen.
Ponieważ początkowa reakcja fosfomutazy jest odwracalna, glukoza 6-fosforanowa może być utworzona z reszt glukozy 1-fosforanu podzielonego z glikogenu. W wątrobie i nerce, ale nie w mięśniu, istnieje enzym, glukoza-6-fosfataza, zdolna do gromadzenia się na 6-fosforanowej glukozie i przekształcania go w wolną glukozę.
Może ci służyć: fotolizaDefosforylowana glukoza może rozprzestrzeniać się na krew, i w ten sposób glikogenoliza wątrobowa znajduje odzwierciedlenie w wzrostu wartości glukozy we krwi (glikemia).
Regulacja syntezy i degradacji
Syntezy
Proces ten jest wykonywany na dwóch podstawowych enzymach: glikogenu syntezazy i glikogenu fosforylazy, tak że gdy jeden z nich jest aktywowany, drugi jest w stanie nieaktywnym. Ta regulacja zapobiega podejmowaniu przeciwnych reakcji syntezy i degradacji.
Aktywna postać i nieaktywna postać obu enzymów jest bardzo różna, a interkonwersja aktywnych i nieaktywnych form fosforylazy i syntetycznego glikogenu podlega ścisłej kontroli hormonalnej.
Adrenalina jest hormonem uwalnianym ze szpiku nadnerczy, a glukagon jest kolejnym, który występuje w hormonalnej części trzustki. Endokrynowa trzustka wytwarza insulinę i glukagon. Islety Langerhans α są tymi, które syntetyzują glukagon.
Adrenalina i glukagon to dwa hormony uwalniane, gdy energia jest potrzebna w odpowiedzi na spadek poziomu glukozy we krwi. Te hormony stymulują aktywację glikogenu fosforylazy i hamują syntezazę glikogenu, stymulując w ten sposób glikogenolizę i hamując glikogenezę.
Podczas gdy adrenalina wywiera działanie na mięśnie i wątrobę, glukagon działa tylko na wątrobę. Te hormony są połączone z specyficznymi receptorami błonowymi w białych komórkach, które aktywuje adenil cyklasowy.
Aktywacja adenylanu cyklazy rozpoczyna wodospad enzymatyczny, który z jednej strony aktywuje białko -białko -białe kwadratazę zależną od AMPC, która nieaktywna do syntetycznego glikogenu i aktywuje fosforylazę glikogenu przez fosforylację (odpowiednio bezpośrednio i pośrednio).
Mięsień szkieletowy ma kolejny mechanizm aktywacji glikogenu fosforylazy przez wapń, który jest uwalniany w wyniku depolaryzacji błony mięśniowej na początku skurczu.
Degradacji
Opisane powyżej enzymatyczne wodospady kończą się na zwiększaniu poziomu glukozy, a gdy osiągają określony poziom, glikogeneza jest aktywowana, a glukogenoliza jest hamowana, również hamując dalsze uwalnianie adrenaliny i glukagonu.
Glikogenezę jest aktywowana przez aktywację fosfatazy fosforylazy, enzymu, który reguluje syntezę glikogenu za pomocą kilku mechanizmów, które sugerują inaktywację fosforylazy kinazy i fosforylazy α, która jest syntezyjnym inhibitorem glikogenu glikogenu.
Insulina promuje wejście glukozy do komórek mięśniowych, zwiększając poziom glukozy 6-fosforanu, który stymuluje defosforylację i aktywację glikogenu syntesazowego. W ten sposób zaczyna się synteza, a degradacja glikogenu jest hamowana.
Funkcje
Glikogen mięśni stanowi rezerwę energii dla mięśni, która, podobnie jak tłuszcze rezerwowe, pozwala mięśniom pełnić swoje funkcje. Będąc źródłem glukozy, glikogen mięśniowy jest stosowany podczas ćwiczeń. Te zastrzeżenia rosną wraz z treningiem fizycznym.
W wątrobie glikogen stanowi również ważne źródło rezerwowe zarówno dla funkcji narządu, jak i dla wkładu glukozy w reszcie ciała.
Ta funkcja glikogenu wątroby wynika z faktu, że wątroba zawiera glukozę 6-fosfatazy, enzym zdolny do wyeliminowania grupy fosforanu glukozy 6-fosforanowej i przekształcania go w wolną glukozę. Wolna glukoza, w przeciwieństwie do fosforylowanej glukozy, można rozprzestrzeniać przez błonę hepatocytów (komórki wątroby).
Może ci służyć: sporulacja: w roślinach, w grzybach i w bakteriachW ten sposób wątroba może zapewnić glukozę do krążenia i utrzymywać stabilny poziom glukozy, nawet w przedłużonych warunkach postu.
Ta funkcja ma ogromne znaczenie, ponieważ mózg jest odżywiony prawie wyłącznie z glukozy we krwi, więc ciężka hipoglikemia (bardzo niskie stężenie glukozy we krwi) może powodować utratę wiedzy.
Powiązane choroby
Choroby związane z glikogenem otrzymują ogólną nazwę „chorób magazynowania glikogenu”.
Choroby te stanowią grupę dziedzicznych patologii charakteryzujących się depozytem w tkankach nienormalnych ilości lub rodzajów glikogenu.
Większość chorób magazynowania glikogenu jest spowodowana genetycznym deficytem natury każdego z enzymów zaangażowanych w metabolizm glikogenu.
Są one podzielone na osiem rodzajów, z których większość ma swoje własne nazwy, a każdy z nich jest wytwarzany przez inny deficyt enzymatyczny. Niektóre są śmiertelne na bardzo wczesnym etapie życia, podczas gdy innym towarzyszy osłabienie i deficyt mięśni podczas ćwiczeń.
Znakomite przykłady
Niektóre z najbardziej widocznych chorób związanych z glikogenem są następujące:
- Choroba von Gierke lub choroba magazynowania glikogenu typu I, jest wytwarzana przez 6-fosfatazowy deficyt glukozy w wątrobie i nerkach.
Charakteryzuje się nieprawidłowym wzrostem wątroby (wątroby) z powodu przesadnej akumulacji glikogenu i hipoglikemii, ponieważ wątroba nie jest w stanie zapewnić glukozy do krążenia. Pacjenci z tym stanem mają zmiany wzrostu.
- Choroba Pompe lub typu II wynika z deficytu α (1 → 4) -glucano 6-glikozylotransferas w wątrobie, serca i mięśniach szkieletowych. Ta choroba, podobnie jak Andersen lub typ IV, jest śmiertelna przed dwoma latami życia.
- Mcardle lub choroba typu V ma deficyt fosforylazy mięśniowej i towarzyszy jej osłabienie mięśni, zmniejszona tolerancja wysiłku, nieprawidłowa akumulacja glikogenu mięśniowego i brak mleczanu podczas ćwiczeń podczas ćwiczeń.
Bibliografia
- Bhattacharya, k. (2015). Badanie i zarządzanie chorobami przechowywania glikogenu wątrobowego. Pediatria translacyjna, 4(3), 240-248.
- Dagli, a., Sentner, c., I Weinstein, D. (2016). Choroba przechowywania glikogenu typu III. Recenzje genów, 1-16.
- Guyton, a., & Hall, j. (2006). Podręcznik fizjologii medycznej (11 wyd.). Elsevier Inc.
- Mathews, c., Van Holde, K., & Ahern, K. (2000). Biochemia (3 wyd.). San Francisco, Kalifornia: Pearson.
- McKiernan, s. 1. (2017). Patobiologia wątroby w magazynowaniu glikogenu. CURR Pathobiol Rep.
- Murray, r., Bender, d., Botham, k., Kennelly, s. 1., Rodwell, v., I Weil, p. (2009). Ilustrowana biochemia Harpera (28. wyd.). McGraw-Hill Medical.
- Nelson, zm. L., & Cox, m. M. (2009). Zasady biochemii lehninger. Omega Editions (Ed.).
- Rawn, J. D. (1998). Biochemia. Burlington, Massachusetts: Neil Patterson Publishers.
- Tarnopolsky, m. DO. (2018). Myopatie związane z zaburzeniami metabolizmu glikogenu. Neuroterapeutyki.
- « Historia argonu, struktura, właściwości, użycia
- Funkcja bijekcyjna Czym jest, jak to się dzieje, przykłady, ćwiczenia »