

Prions
- 2935
- 328
- Herbert Wróblewski
Prionki są słabo złożonymi białkami, które przekazują tę błędne informacje na inne białka i powodują przenoszone gąbczaste encefalopatie. Źródło: Wikimedia Commons Co to są prions?
prions Są to białka bez genomu lub kwasów nukleinowych, które działają jako środki zakaźne. Znajdują się w normalnej błonie komórkowej, tylko jako słabo złożone białka i/lub z nieprawidłową trójwymiarową strukturą.
Te białka są odpowiedzialne za wielokrotne choroby zwyrodnieniowe i bardzo wysoką śmiertelność, które wpływają na tkanki nerwowe i strukturę mózgu.
Nazywa się je również chorobami prionowymi. Jednym z najważniejszych, które wpływają na ludzi, są Kuru, choroba Gerstmann-Sträussler-Scheinker, zespół Creutzfeldt-Jakob i śmiertelna rodzina bezsenność.
Charakterystyka prionka
- PRONS to struktury białkowe obecne w błonach komórkowych. Białka te mają zmienioną formę lub konformację [PRP (SC)].
- W odniesieniu do jego mnożenia, osiąga się go przez konwersję form, jak w przypadku drżących choroby. W tej chorobie prions rekrutuj PRP (C) (białka prionowe o konformacji bez opieki) do stymulowania konwersji do izoformu PRP (SC).
- Te niezwykłe białka zdolne do rozprzestrzeniania się, nie mają kwasów nukleinowych. Dowód jest taki, że są one odporne na promieniowanie x i promieniowanie ultrafioletowe. Środki te łatwo rozkładają kwasy nukleinowe.
- Białka prionowe, z których składa się Prions (PRP), znajdują się w całym ciele, nie tylko ludzi, ale także innych zdrowych kręgowców.
- Niektóre badaczy udało się wykazać, że u myszy białka te aktywują naprawę mielinową w komórkach obwodowych układu nerwowego. Wykazano również, że brak tych powoduje demielinizację takich komórek nerwowych.
Struktura prion
Wiedza na temat struktury prionów znajduje się głównie w badaniach przeprowadzonych w bakteriach Escherichia coli.
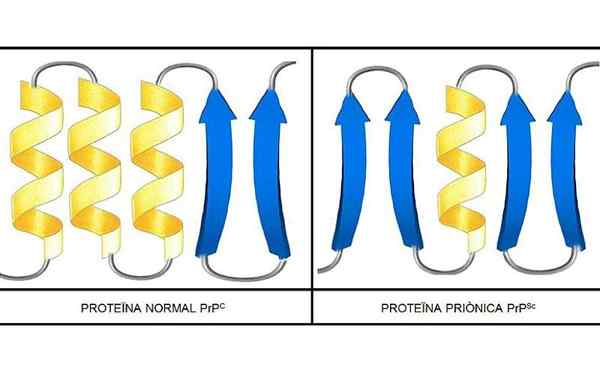
Badania przeprowadzono, że polipeptydy w łańcuchowym PRP (C) (normalne) i PRP (SC) (zakaźne) są identyczne w składzie aminokwasów, ale różnią się w konformacji 3D i fałdowaniu ich.
PRP (c)
Te nieinfekcyjne prions obecne, u ludzi, 209 aminokwasów. Mają link disiarczkowy. Jego struktura jest alfa-helikoidalna, co oznacza, że ma spiralne aminokwasy (śmigła alfa) i kilka płaskich pasm aminokwasowych (liście beta).
Może ci służyć: Nauk o biologii pomocniczejBiałka tego nie można oddzielić przez wirowanie, co oznacza, że nie jest ono osadowe. Jest łatwo trawiony przez szeroką proteazę szerokiego spektrum zwanego proteinazą k.
PRP (SC)
Jest to zakaźne białko, które przekształca PRP (C) w zakaźne izoform PRP (SC).
Niewiele wiadomo na temat jego struktury 3D, jednak wiadomo, że ma niewiele helikalnych kształtów i więcej płaskich pasm lub liści beta. Zmiana w kierunku izoform jest tak zwana fundamentalne zdarzenie chorób prionowych.
Funkcje prionowe
Białka prionowe komórkowe [PRP (C)] znajdują się na powierzchni komórki szerokiej gamy narządów i tkanek. Niewiele wiadomo na temat fizjologicznych funkcji prionów w ciele.
Mimo to doświadczenia wykonane u myszy wskazują na możliwe funkcje, takie jak:
Z metabotropowymi receptorami glutaminianowymi
Wykazano, że PRP (C) działa z receptorami glutaminianowymi (jonotropiki i metabotropy). PRP (C) uczestniczy jako receptor synaptotoksycznych oligomerów peptydu komórkowego powierzchni komórkowej.
W rozwoju embrionalnym
U myszy rodzinnych Murinae odkryto, że białka prionowe PRP (c) są wyrażane kilka dni po implantacji, w rozwoju embrionalnym.
Wskazuje to, że odgrywają rolę podczas rozwoju tych małych ssaków. Papier, który według naukowców jest związany z regulacją neuritogenezy (produkcja aksonów i dendrytów neuronów).
Działają również w wzroście aksonów. Te białka prionowe są nawet zaangażowane w rozwój obwodu móżdżku. Z tego powodu uważa się, że brak tych prionów PRP (c) pociąga za sobą opóźnienie w rozwoju motorycznym gryzoni.
Neuroprotektor
W badaniach nad nadekspresją PRP (C) z powodu orientacji genów stwierdzono, że brak tych prionów powoduje problemy z nawadnianiem krwi do niektórych miejsc w mózgu (ostre niedokrwienie mózgu).
Oznacza to, że białka prionowe działają jako neuroprotektory. Dodatkowo wykazano, że nadekspresja PRP (C) może zmniejszyć lub poprawić zmiany spowodowane niedokrwieniem.
Obwodowego układu nerwowego
Niedawno odkryto fizjologiczną funkcję PRP (C) w utrzymaniu szpiku peryferyjnego.
Może ci służyć: dystrofina: charakterystyka, struktura i funkcjePodczas badania laboratoryjnego odkryto, że przy braku białka prionowego myszy laboratoryjne rozwinęły niedobory nerwów, które przenoszą informacje z mózgu i rdzenia kręgowego, w tak zwanej neuropatii obwodowej.
Śmierć komórki
Istnieją niektóre białka prionowe i znajdują się w innych częściach ciała inne niż mózg.
Funkcje takich białek polega na inicjowaniu, regulacji i/lub kontrolowaniu śmierci komórki, gdy ciało jest atakowane (na przykład przez sulony), zapobiegając w ten sposób propagacji patogenu.
Ta szczególna funkcja tych białek sprawia, że naukowcy myślą o możliwym znaczeniu nieinfekcyjnych prionów w walce z patogenami.
Pamięć długoterminowa
Badanie przeprowadzone w Stowers Institute, w Missouri, EE. Uu., Wykazał, że prony PRP mogą pełnić funkcję w utrzymaniu długoterminowej pamięci.
Badanie wykazało, że niektóre białka prionowe mogą być kontrolowane w celu utrzymania funkcji fizjologicznych pamięci długoterminowej.
Odnowienie komórki matki
Badanie białek prionowych wyrażanych w komórkach tkankowych macierzystych, ujawniło, że wszystkie te komórki macierzyste (hematopoetyczne), wyrażają białka prionowe w błonie komórkowej. Uważa się więc, że uczestniczą w złożonym i bardzo ważnym procesie odnawiania komórek.
Choroby spowodowane przez prion
Najczęstsze choroby prionowe to:
Choroba Creutzfeldt-Jakob (ETJ) (ECJ)
Uważana za najczęstszą chorobę prionową wśród ludzi, jest to patologia kosmopolityczna, czyli na całym świecie rozmieszczenie. Może wystąpić dziedzictwo (rodzina), sporadyczne lub zakaźne.
Choroba Gerstmann-Sträussler-Scheinker
Jest to choroba spowodowana prionami w odziedziczonym zakaźnym lub autosomalnym dominującym procesie encefalicznym. Choroba objawia się u osób od 40 do 60 lat.
Prionopatia o zmiennej wrażliwości proteazy
Jest to bardzo rzadka choroba, do tego stopnia, że jej zakres występowania wynosi od 2 do 3 przypadków na 100 milionów mieszkańców. Patologia jest podobna do choroby Gerstmann-Sträussler-Scheinker.
Śmiertelna bezsenność
Jest to dziedziczna lub znana choroba, chociaż może również wystąpić sporadycznie. Wiadomo, że choroba jest spowodowana dominującą mutacją dziedziczną lub autosomalną.
Może ci służyć: gatunki endemiczneKuru
Ta choroba prionowa została wykryta tylko u mieszkańców Papui Nowej Gwinei. Jest to choroba związana z kanibalizmem i kulturową tradycją obrzędu pojedynków dla umarłych, gdzie ci ludzie jedzą ludzki mózg.
Choroby u zwierząt
Wśród patologii wytwarzanych przez prion u zwierząt jest bydlę encefalopatii gąbczastej. Ta choroba sędziła spustoszenie w Europie, w zdrowiu publicznym, zwierząt i gospodarki dotkniętych krajów.
Inne choroby u zwierząt to złom, przenoszona encefalopatia norki, przewlekła choroba zużycia (w jeleniu) i encefalopatia gąbczastej koci.
Choroby te, podobnie jak te przedstawione u ludzi, nie mają skutecznego leczenia, więc zapobieganie ma zasadnicze znaczenie, szczególnie po zakaźności u ludzi, które wystąpiły z powodu spożywania mięsa zakażonych krów.
Zabiegi
Do tej pory nie wiadomo żadne lekarstwo na choroby prionowe. Leczenie jest objawowe. Pacjentom zaleca się planowanie opieki paliatywnej i analizy genetycznej oraz porady dla członków rodziny.
U pacjentów z chorobami prionowymi, takimi jak antywirusowe, przeciwnowotworowe.
Jednak obecnie nie ma dowodów wskazujących, że niektóre z tych objawów lub poprawiają przeżycie chorych.
Zapobieganie
PRONS są odporne na różne zmiany fizyczne i chemiczne. Jednak stosuje się różne techniki, aby uniknąć zanieczyszczenia pacjentów z zanieczyszczonymi instrumentami chirurgicznymi.
Jednym z najczęściej stosowanych technik jest sterylizacja sprzętu w autoklawie w 132 ° C przez godzinę.
Z drugiej strony Światowa Organizacja Zdrowia (WHO) opracowała środki w celu uniknięcia rozprzestrzeniania się chorób prionowych. Ta organizacja ustanawia zasady zarządzania zakazanymi lub potencjalnie ryzykownymi tkankami, takimi jak: oczy, mózg, jelito, migdałki i rdzeń kręgowy.
Bibliografia
- Prion, środek zaraźliwy. Odzyskane z Britannica.com.
- Co to jest prion? Odzyskane z naukowego.com.
- Prion. Odzyskane z.Wikipedia.org